I worked to launch a research project involving the use of spectroscopic techniques to quantify nanoplastics at varied interfaces/complex settings and creating methods for labeling nanosized polymers with fluorescent dyes to employ them in single molecule studies.
Institution Georgia State University Department of Chemistry
Timeline 8 Months
Role Undergraduate Student Researcher
Date Completed May 2023
Summer 2022 Research Externship
This summer I have the opportunity to do research in the field of computational and quantum chemistry ⚛️ . Continue reading below and/or view my three nugget video presentation to learn more! ⬇️
Practice Session of 3-Minute Nugget Video Presentation Explaining My Research
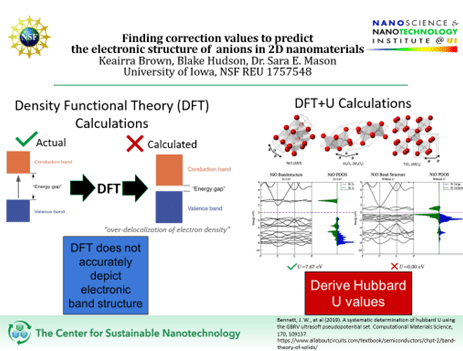
Project Title: Finding correction values to predict the electronic structure of anions in 2D nanomaterials
Density Functional Theory (DFT) is a computational tool that allows us to predict the electronic and physical properties of solid-state materials such as nanomaterials like complex metal oxides. To clarify, DFT allows us to guess the state and behavior of electrons in nanomaterials, meaning we can describe the state of these negatively charged particles known as electrons in nanomaterials. In other words, we can describe their state of electrons in terms of energy or momentum, which has resulted in many discoveries. DFT is awesome! It is a popular and quick method that is used amongst computational chemists and even quantum physicists, which has opened doors to identifying new materials and has even helped scientists develop new design principles that have for instance led to the discovery of new and improved lithium-ion battery (LIB) cathode materials. Moreover, screening of these metal oxide materials using DFT has led to the discovery of cheaper and more stable candidates to improve LIBs.
As mentioned before, DFT is awesome—some may even call it SUPER! However, like Superman, it has its very own kryptonite. For transition metals with electrons in 3d, 4d- and even 4f orbitals, it fails to accurately depict the state of electrons in available energy levels where electrons move freely. Simply, DFT is known to predict the electron band structure of these metals incorrectly, describing these metals as conductors when they are not.
To remedy this limitation, the Mason lab strives to identify optimal correction values known as Hubbard U correction values that help to fence in these d and f orbital electrons to keep them from moving freely which corrects electronic band structure.
In previous research, this was attained by finding Hubbard U values for three different crystal structures of metal oxides, rock salt, corundum, and rutile. It was found that the U values discovered for each metal oxide gave a pretty accurate depiction of their electron band structure and were close in range that an average of the values could be used.
Currently we are working to substitute the anions of four crystal structures: rock salt, corundum, rutile, and wurtzite with semiconductors to create a database of Hubbard U values for 2D nanomaterials
Company The NSF Center for Sustainable Nanotechnology